解决方案-ICH Q3C和Q3D在中国药典的转化实施:修订通则0861和新增通则0862
修订0861和增订0862元素杂质通则是ICH Q3C和Q3D在中国药典转化实施的重要内容之一,与之充分协调。用基于风险的方法对残留溶剂和元素杂质进行评估,对药品全生命周期内进行控制,同时对凡例修订也产生影响。
药品中的残留溶剂系指在原料药或辅料的生产中,以及在制剂制备过程中使用的,但在工艺过程中未能完全去除的有机溶剂。当药品所含的残留溶剂水平高于安全值时,就会对人体或环境产生危害,应尽可能除去所有残留溶剂,以符合制剂质量标准、生产质量管理规范(GMP)或其他质量要求。
| 2020年版ChP | 2025年版ChP | 修订说明汇总 |
|---|---|---|
| 0861 残留溶剂测定法 | 0861残留溶剂 | 修订通则名称 |
| 定义:是指在原料药或辅料的生产中,以及在制剂制备过程中使用的,但在工艺过程中未能完全去除的有机化合物。 | 定义: 是指在原料药、辅料或制剂生产过程中使用或产生的,并在实际工艺过程中不能完全出去的有机挥发性化合物 | 有修订,同ICH Q3C定义协调 |
| / | 评估与控制的一般原则 | 新增,引入Q3C的内容,本通则还强调:由于不同生产企业或不同工艺所用溶剂种类可能存在差异,无论各品种正文中是否设置残留溶剂检查项,生产企业均应基于本通则的要求,遵循质量风险管理原则,结合生产工艺,对使用或可能产生的残留溶剂,采用本通则推荐的方法或其他经验证、核准的方法进行测定和控制,使残留溶剂量符合本通则规定的限度要求。 |
| / | 评估与控制:基于风险评估的残留溶剂分类 | 新增,协调Q3C的内容 |
| / | 评估与控制:残留溶剂的限度 | 新增,协调Q3C的内容,强调表中列出的溶剂分类及其建议限度来源于ICH Q3C,且将随安全性数据更新和ICH Q3C版本的更新而变更 |
| / | 评估与控制:第二类溶剂限度的确定方法 | 新增,协调Q3C的内容 |
| / | 评估与控制:残留溶剂的报告方式 | 新增,协调Q3C的内容 |
| 测定方法 | 测定方法 | 新增概述,药品中残留溶剂的鉴别、限度检查和定量测定通常采用色谱技术如气相色谱法。可采用本通则推荐的方法,或选择与药品生产特定情况相适应的经验证的分析方法。当仅有第三类溶剂存在时,也可采用经适当验证的非专属性方法进行检查,如干燥失重检查法。验证时应考虑溶剂挥发性对分析方法的影响。 |
| 色谱柱 | 色谱柱 | 少量文字修订 |
| 供试品溶液的制备 | 供试品溶液的制备 | 有修订 |
| 对照品溶液的制备 | 对照品溶液的制备 | 有修订 |
| 测定法(第一法、第二法、第三法) | 测定法(第一法、第二法、第三法) | 有修订 |
| 系统适用性试验 | 系统适用性试验 | 理论塔板数:删除原具体数值要求, 补充了在柱效影响分离效能时,可规定色谱柱应达到的最小理论板数。 分离度:删除具体要求,增加必要时可用峰谷比描述 增加了对称性、灵敏度、重复性相关内容的描述 |
| / | 残留溶剂的鉴别 | 增加内容,提供两种鉴别方法,保留时间删除了RART列表 强调两个保留时间不同的色谱峰归属于不同化合物,但两个保留时间一致的色谱峰有时未必可归属为同一化合物,在作未知物定性分析时应特别注意。 |
| / | 残留溶剂的检查和定量 | 原通则0861的“计算法”的内容整合至“残留溶剂的检查和定量”中,增加了对第三类溶剂的测定。 列出两种方法:限度检查内标法(峰面积比)外标法(峰面积),定量测定按外标法或内标法计算残留溶剂的量。特别注意对第三类溶剂的鉴别根据需要进行检查。另外结合残留溶剂的测定,给出分析策略图。 |
| 附注 | 分析方法建立和使用中的其它考虑(1)~(10) | 对原“【附注】内容进行整理,删除了利用保留值定性的内容,增加了(5)和(6)。 (1)采用等温法和程序升温法的考虑,无修订 (2)顶空条件选择的考虑,将平衡时间由30-45分钟修订为30-60分钟,新增对平衡温度的考虑 (3)沸点较高的残留溶剂的测定,删除顶空的相关描述 (4)含氮的碱性残留溶剂的测定,将“化合物” 修订为“残留溶剂” (5)新增,含羧酸的酸性残留溶剂的测定 (6)新增,流速的选择 (7)检测器选择的考虑,无修订 (8)干扰峰的排除,少量文字修订 (9)定量方法的验证,无修订 (10)仲裁方式,以内标法或标准加入法的结果为准,无修订 |
| 附表(1-3) | 表1-4 | 删除了原附表1-3 根据ICH Q3C的溶剂分类,新增了表1~表4,内容包括溶剂的分类、各溶剂的PDE值、限度(ppm)以及CAS号等信息 |
岛津残留溶剂检测应对方案

| 产品 | 固定相 | 适合范围 |
|---|---|---|
| SH-5 | 5%二苯基/95%二甲基聚硅氧烷 | 磺酸酯类基因毒性杂质、多环芳烃、环氧乙烷、高沸点残留溶剂杂质 |
| SH-I-624Sil MS | 6%氰丙基苯基/94%二甲基聚硅氧烷 | 乙醇挥发性杂质、残留溶剂(优选)、环氧乙烷 |
| SH-PolarWax | 聚乙二醇 (PEG) | 脂肪酸组成、亚硝胺类、磺酸酯类基因毒性杂质、醇酯等极性较强残留溶剂分析、极性原料溶剂纯度分析 |
| SH-Volatil Amin | 碱性化处理的100%二甲基聚硅氧烷 | 胺类溶剂残留分析,同时可应用于醇类、游离酸、酯、胺类混合检测 |
| SH-PolarD | 硝基对苯二甲酸改性的聚乙二醇 | 酸性溶剂残留分析,游离酸、醇、酯类 |
| Shim-pack Scepter C18 | 十八烷基硅烷 | 液相方法通用柱,耐受pH 1-12,可用于分析酸性残留溶剂 |
|
备注:含羧酸的酸性残留溶剂测定(如甲酸、醋酸等),采用气相色谱法测定一般响应值较低,可通过酯化衍生等样品处理方式提高检测灵敏度,也可采用液相色谱法或其它适宜的方法测定。采用液相色谱法测定时,流动相和供试品溶液应呈酸性。 |
||
存在于药品中的元素杂质有多种来源,生产过程中使用的原料药、辅料、生产设备、水和包装材料等均可能引入元素杂质,贮存过程中包装材料中的元素杂质还可能发生迁移而被引入药品中。这些元素杂质可能是有意添加引入(如原料药或辅料合成过程中有意添加的催化剂残留),也可能是无意引入(如与生产设备或包装系统相互作用产生的杂质或药品各个组分中存在的杂质)。为了治疗作用而有意添加到药品中的元素不属于元素杂质。因元素杂质不能为患者提供任何治疗作用,某些元素杂质甚至有一定毒性,所以它们在药品中的量需被控制在可接受的限度范围内。以符合制剂质量标准、生产质量管理规范(GMP)或其他质量要求。
| 通则0862内容 | ICH Q3D(R2) | 对比说明 |
|---|---|---|
| 前言(未实际列出标题,包括元素杂质来源、定义、目的及适用范围的描述) | (1)前言(5.2)元素杂质的潜在来源 (2)范围 |
基本一致 |
| 1、风险评估中建议考察的元素 | (4)元素分类 (5.4)风险评估建议考虑的元素 (附录5)皮肤和透皮给药途径的元素杂质的限度 |
一致 |
| 2、形态 | (8)形态和其他考虑 (附录3)有关砷和汞的部分内容 |
一致 |
| 3、每日允许暴露量 | (3.4)注射剂 (附录2)元素杂质的既定PDE值 (附录5)皮肤和透皮给药途径的元素杂质的限度 |
一致 |
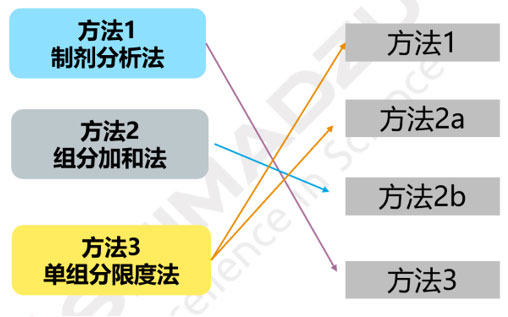
| 4、限度确认方法 | (7)PDE值与浓度限度的相互转换 | 有差异,但与ICH Q3D方法对应,如下图 |
 |
||
| 5、评估 | (5.5)评估 | 一致 |
| 6、控制阈值 | (5.6)风险评估过程总结 | 一致,将30%PDE(以及Ni和Co的CTCL)定义为控制阈值,作为元素杂质水平显著性的衡量指标,可用于判断药品中的元素杂质是否需要额外控制 |
| 7、元素杂质的控制 | (6)元素杂质的控制 | 一致,与检验标准有关的内容:建立原料药、辅料或物料(如合成中间体)、制剂的元素杂质标准限度;对药品中元素杂质进行定期检测 |
| 8、测定方法 | (9)分析方法 | 0862提供具体程序 |
岛津元素杂质检测应对方案
有关测定方法:通则0862明确指出,任何可以满足质量控制要求的方法均可用于元素杂质的测定。检测方法包括但不限于电感耦合等离子体质谱法(通则0412)、电感耦合等离子体原子发射光谱法(通则0411)原子吸收分光光度法(通则0406)、X射线荧光光谱法(通则0461)、重金属检查法(通则0821)、检查法(通则0804)砷盐检查法(通则0822)等。上述方法通常测定的是不同形态元素的总量,若需区分元素的形态,可采用电感耦合等离子体质谱法与分离技术如液相色谱法联用的方法,或其他适宜的方法进行测定。